- Review
- Open access
- Published:
Resistance to cancer chemotherapy: failure in drug response from ADME to P-gp
Cancer Cell International volume 15, Article number: 71 (2015)
Abstract
Cancer chemotherapy resistance (MDR) is the innate and/or acquired ability of cancer cells to evade the effects of chemotherapeutics and is one of the most pressing major dilemmas in cancer therapy. Chemotherapy resistance can arise due to several host or tumor-related factors. However, most current research is focused on tumor-specific factors and specifically genes that handle expression of pumps that efflux accumulated drugs inside malignantly transformed types of cells. In this work, we suggest a wider and alternative perspective that sets the stage for a future platform in modifying drug resistance with respect to the treatment of cancer.
Background
In US only, the newly diagnosed cancer patient is 1,665,540 every year and the estimated death is 585,720 [1] which are increasing as countries become more developed and more people reach advanced ages. Therefore, many efforts are being done in the war against cancer [2]. One of these efforts is the continuous development of new drugs and forms of chemotherapy.
In cancer, chemotherapy represents the backbone of treatment for many cancers at different stages of the disease. Therefore, chemotherapeutic resistance results in therapeutic failure and usually, (eventually) death. To address these limitations, many researchers focus on how cancer cells manipulate their genomes and metabolism to prevent drug influx and/or facilitate efflux of accumulated drugs, the so called: “the neostrategy of cancer cells and tissues” [3]. In this work, we show that drug resistance is a multifactorial phenomenon that requires attention to the host as well as the tumor and that such factors are organized at different levels (Figure 1).

Diagrammatic representation describes that cancer chemotherapy resistance is a multi-factorial phenomenon that could be organized as multilevel structure.
Macroscopic (systemic) resistance [host–related factors]
One of the major effects of host-related factors that determine the activity of the drug is pharmacokinetic. Pharmacokinetics is defined as the action of the body in response to drug and can be divided into the following consecutive steps: Absorption, Distribution, Metabolism and Excretion (ADME) [4]. Here we introduce the concept of “Pharmacokinetic Resistance” to describe the body-related factors that alter the effectiveness of the drug, so that it either does not reach its target and/or cannot accomplish its intended goal. Chemotherapeutics must be in contact with the tumor.
Absorption
There are growing evidences suggesting that orally cancer chemotherapy is preferable to intravenous administration, because: (1) it is low-cost from the national health services perspective (i.e. does not require hospitalisation), (2) it can increase the drug’s antitumor activity by prolonging it’s time to clearance [5], (3) it reduces drug toxicity [6], (4) increase patients’ compliance and improve pharmacoeconomic issues [7–9]. However, to maintain a sufficient amount of orally administered chemotherapeutics, several factors should be taken into consideration:
P-gp
Permeability glycoprotein (P-gp), also known as multidrug drug resistance protein (MDR) is found along the gastrointestinal tract (GIT) [10], including the small intestine as primary site for the epithelial absorption of many orally administered drugs [11]. It has been shown that P-gp reduces the oral bioavailability of some anticancer drugs [12]. Concomitant administration of some of the antineoplastic agents leads to the overexpression of P-gp that results in bioavailability reduction of several these agents, e.g. Imatinib [13]. P-gp expression, in the gut, is a subject of interindividual variation due to either genetic polymorphism or pathologic condition [14, 15] and so fluctuates the bioavailability of several antineoplastic agents e.g. paclitaxel [12].
Food
The effects of food on drug absorption and bioavailability have been attracting attention very much and stimulated a long debate of whether fasting improved or worsened a drug’s bioavailability [16–18]. It has been shown that the half-life period of orally applied Topotecan is longer than that of intravenous administration. The administration of Topotecan with a high-fat breakfast shows a small decrease in the absorption rate but does not affect the extent of the absolute absorption [19]. St John’s wort, induces the expression of Pregnane X receptor, a xenobiotic or detoxification sensor, which reduces the efficacy of some antineoplastic agents, e.g. Irinotecan [4]. CYP3A4 is a metabolising enzyme (see below) which has been found in the intestine presumably as a defense strategy against xenobiotics [20]. It is well known that grapefruit juice abates the presence of CYP3A4, which is beneficial for the application of antineoplastic oral agents with low bioavailability [21]. Thus, it becomes apparent that the interaction between food and antineoplastic agents should be cautiously monitored to maintain a sufficient bioavailability [22–24].
Distribution
The “volume of distribution” (Vd) of the drug is a hypothetical volume that outlines drug distribution into tissues [25, 26]. A higher Vd means that more of a drug penetrates into a tissue while it is more diluted (present at lower concentrations) in the plasma. The distribution of the drug between plasma and tissues relies on several factors. Some of them include:
-
1.
Gender Metronidazole, a bactericide and protozoocide, also displays a potential activity as an anticancer drug, especially as a radiosensitizer in hypoxic regions [27], and shows a lower Vd in women [28] Furthermore, women are subject to pharmacokinetic variability during their menstrual cycle [29]. Therefore, it is critical to consider the gender factor also to age if the concentration of the drug in the plasma is to be determined or needs to be defined.
-
2.
Weight Weight is a paramount factor that determines dose adjustment [30]. Cancer patients often lose weight during tumor progression [31–33]. Therefore, dosing based on body weight should be routinely re-adjusted to the current weight [34, 35].
-
3.
Plasma proteins when a drug is available in the blood (plasma), the drug will be found either in free or bound form to plasma proteins (see Figure 2). Albumin and Alpha 1-acid glycoprotein are an example of drug-binding plasma proteins. While albumin carries acidic drugs, the alpha 1-acid glycoprotein will carry basic and neutral lipophilic drugs [36–38].
Figure 2 -
Albumin: Albumin is an important binding protein in the blood. It is a powerful prognostic indicator reflecting diseases’ severity [39, 40], and its prognostic value is subject to gender differences [41]. It has been shown that etoposide is subject to individual variations (Population diversity) changes in the albumin serum concentration and/or age [42, 43].
-
Alpha-1-acid glycoprotein (AGP or AAG) sometimes called orosomucoid (ORM) is an acute phase plasma alpha globulin glycoprotein. AGP is a critical determinant factor for the activity of several anticancer agents, e.g. imatinib [44]. Variation of AGP’s serum concentration also affects the anticancer activity of Gefitinib [45]. Wu et al. showed that the γ-secretase inhibitor RO4929097 (Notch signaling blocker) is bound in plasma with high affinity to AGP and can be competitively replaced by GDC-0449 (Hedgehog inhibitor). This consequently increases the availability of potentially active RO4929097 [46]. Therefore, it was suggested that AGP monitoring is critical to predict the pharmacodynamics response to a combined RO4929097/GDC-0449 treatment [46].
-
-
4.
Circadian rhythm The circadian timing system comprises peripheral oscillators located in most tissues of the body and a central pacemaker located in the suprachiasmatic nucleus (SCN) of the hypothalamus [47]. The circadian rhythm has been implicated in the pathophysiology of several diseases [48–50], drug action [51, 52] and pharmacokinetics of drugs as well [53–55]. Plasma protein levels reach their minimum around 4:00 a.m. and start to increase around 8:00 a.m. [56]. This circadian rhythm can be masked at younger ages, but it aggravates and becomes clearer with aging [56]. Therefore, proper dosage timing should result in higher drug concentrations reaching the tumor site. It has been shown that alpha one acid glycoprotein (AGP) is subject to circadian rhythms [57]. In cancer, it has been demonstrated that Cisplatinum shows a considerable variability in its binding capacity day and night [58, 59]. Thus, circadian rhythmicity has a significant impact on a drug’s pharmacophore i.e. the active site of the drug molecule, and a delicate balancing between chronotolerability (minimum toxicity to host) and chronoefficacy (maximum cytotoxicity) is required [60]. Moreover, most of currently used anticancer agents act against highly proliferating cells, and since the basal metabolic rate is increased at night, it seems adequate to administer anticancer drugs at night instead of during the day.

Metabolism
Drug metabolism does not reflect the conventional metabolic pathways, i.e. anabolic for biomass or catabolism for energy production. Instead, drug metabolism involves changing drug polarity and thus its hydrophilicity, to facilitate its excretion from the body. Drug metabolism occurs through two steps: the first involves reactions such as hydroxylation or oxidation [61, 62] of lipophilic drugs to make it vulnerable to the addition of glutathione, glucouronic acid or an amino acid [63, 64]. Although it is generally accepted that drug metabolism is a biological strategy of detoxification, some metabolism enzymes such as cytochrome P450 and glutathione S–transferase could be taken advantage of as they can activate certain anticancer drugs [65].
-
A group of cytochrome P450 (CYP) enzymes responsible for the first step, the introduction of reactive or polar groups into xenobiotic groups [66]. CYP enzymes have been shown to activate some of the anticancer agents [67], as well as inactivate other anticancer drugs [68]. Overexpression of CYP450 in cancer patients might lead to resistance due to the rapid inactivation of the drug. Moreover, the presence of CYP450 shows interindividual variation [69–71] and so its detection, identification, and quantification prior to starting treatment is essential.
-
Glutathione–S–Transferases (GSTs) are endogenous detoxifying enzymes [72] which mediate the second step of drug metabolism [73]. Overexpression of GST correlates with drug resistance [74–76]. This resistance could occur pharmacokinetically by metabolizing the drugs into inactive molecules [77]. Others suggest this resistance corresponds to detoxification via energy-dependent, transporter-mediated efflux of drugs or drug conjugates from the cell [78]. Also, GST generates resistance by suppressing apoptosis through its ROS-scavenging activity [79, 80] or via MAP kinase inhibition [81]. Conversely, GST is involved in the activation of certain drugs such as γ -Glutamyl- α -amino-(2- ethyl-N, N, N,N- tetrakis (2-chloroethyl) phosphoro-diamidate)-sulfonyl-propionyl)-(R)-(-) phenylglycine (TER286) [65], 6-mercaptopurine (6-MP) [82], and TLK-286 [83]. Because GST shows variability in its expression across populations [84–87], GST detection prior to chemotherapy could be utilized to inform established therapeutic strategies. Potential GST inhibitors include ethacrynic acid and buthionine sulfoximine [88]. These agents might be consumed concomitantly to keep GST in check in cancer and during cancer therapy. Interestingly, GST is subject to circadian rhythmicity that also affects the activity of 5–fluorouracil (5-FU) and Oxaliplatin [89–91].
-
Extrahepatic metabolism: Typically, the liver plays a major role in drug metabolism. Drug-metabolizing enzymes are also present at other sites e.g. lung, gut, kidney, urinary bladder, skin [92–95]. Some of anticancer agent is a subjected into extrahepatic metabolism e.g. Oracin [95], and Paclitaxel could be subjected to extrahepatic metabolism too [96]. Extrahepatic metabolism also subjected to interindividual variation [95]. So, this issue should be addressed careful monitoring of these agents should discuss toward individualized chemotherapy [68, 97].
Excretion
Excretion from the body is the final step in drug removal. Commonly, excretion of drugs occurs through two main routes: biliary and renal excretion.
-
Biliary or bile duct excretion: MDR (ABC) mediates biliary excretion of xenobiotics [98]. Overexpression of ABC is correlated with an increase in biliary excretion [99–102]. Therefore, careful monitoring of ABC expression should be taken into account when defined anticancer drug is prescribed for the patients and this anticancer drug is knowingly excreted through the bile.
-
Renal excretion: The kidney is the primary organ by which drugs are excreted. Interindividual renal drug excretion variability might be due to gender differences [103, 104] and ethnic differences [105]. So, changes in the glomerular filtration rate (GFR) have a direct effect on anticancer drug availability.
Drug–drug interactions
Cancer chemotherapy is administered in the form of a cocktail, the combined application of several chemotherapeutics. Such a combination is designed to reduce toxicity and to decrease the likelihood of resistance. One drug alone would require a higher concentration and, so the side effects caused would be increased. Using a combination reduces the side-effects of each single drug as it can be applied at a significantly lower concentration [106]. A tumor consists of a heterogeneous population and it is commonly thought that using a “cocktail” of several agents will target different populations and thus reduce the selective pressure by using single agents (the use of only one single agent might kill one defined population and positively select a pre-adapted one that will remain and grow). Therefore, a combination therapy is useful in treating cancer. Conversely, it needs to be pointed out that co-administration of drugs might result in antagonism such that one drug may counteract or neutralize another one:
-
Agents that target tumor vascularization: tumors require a blood supply for the provision of oxygen and nutrients [107], removal of metabolites [108] and to support metastasis [109]. It is widely assumed that administration of agents that target tumor vasculature (antiangiogenic therapy) eventually interrupts tumor progression. There are two classes of these agents (1) Angiogenesis inhibitors; they inhibit the tumor that has initiated the angiogenic process and (2) vascular disrupting agents that destruct the existing tumor vessels. Those agents might limit perfusion of cytotoxic drugs especially upon chronic administration [110]. Moreover, it is postulated that antiangiogenic therapy is useful in the management of resistance to chemotherapy [111], however, diminishing tumor vascularization may accelerate the adaptation to hypoxia while increasing the necrotic zone by accumulation of metabolites and so worsen tumor prognosis [112]
-
NaHCO3 has been recently used systemically in the treatment of cancer [113]. It induces systemic alkalosis. By elevation of urine pH, methotrexate excretion is greatly enhanced. Therefore, NaHCO3 modulates the pharmacokinetics of methotrexate [114].
-
Tamoxifen is a prodrug that needs to be metabolized to its active form by CYP2D6, CYP3A, CYP2B6 and CYP2C19 [115]. Some drugs, particularly from the group of selective serotonin reuptake inhibitors, inhibit CYP2D6 and so reduce the efficacy of Tamoxifen by decreasing amount of its active metabolites [116–118].
-
Pravastatin, an HMG-CoA reductase inhibitor, is correlated with biliary excretion [119, 120] as a substrate of P-gp [121]. Also, it induces P-gp expression as well [122] and so may promote resistance to other compounds.
Microscopic (local) resistance [tumor related factors]
The loss of ability of drugs to kill cancer cells could also be due to failure at the tumor site. Such disability could occur via several mechanisms. Some of them are:
Evolutionary resistance
Also termed biochemical resistance [123, 124], acquired resistance [66, 125], active resistance [126], or extrinsic resistance [127]. Evolutionary resistance is an ancient type of resistance that can be found in bacteria even prior exposure to antibiotics [128–132]. Evolutionary resistance could occur either through interfering with drug resident time intracellularly and/or altering its site of action.
Alteration of drug residency in cancer cells
There are several proteins that alter drug residency in cancer cells. Some of these include:
P-gp
P-glycoprotein 1 (permeability glycoprotein, abbreviated as P-gp or Pgp) also known as multidrug resistance protein 1 (MDR1) or ATP-binding cassette sub-family B member 1 (ABCB1) or cluster of differentiation 243 (CD243). It is a glycoprotein that in humans is encoded by the ABCB1 gene [133, 134]. Commonly, P-gp is localized at the plasma membrane [135] of colon, jejunum, bile canaliculi, renal tubular cells, placenta, the luminal surface of capillary endothelial cells, testes, pancreas and blood–brain barriers (BBB) [135–138]. P-gp might have a role in the normal secretion of metabolites. P-gp also induces expression of CYP3A4 [139] that in turn may deactivate some anticancer drugs (see Table 1 that shows the pharmacological modulators of P-gp).
In resistant cancer cell lines, P-gp is localized in the Golgi apparatus and the rough endoplasmic reticulum [140, 141]. Also, it is expressed in mitochondrial cristae [142, 143] to protect the accumulation of mitochondria [144] or prevent nuclear accumulation by expression of P-gp at the nuclear envelope [141, 145].
Expression of P-gp fluctuates with elevated expression level in untreated cancer into higher level upon relapse after chemotherapy and undetectable or low level in the expression in drug sensitive tumors [134, 146] which means there is no unifying theorem correlating expression of P-gp and cancer treatment.
MRPs
Multidrug resistance-associated protein MRP1 (ABCC1) was the first of the xenobiotic-transporting MRP-related proteins to be cloned and was identified based on its overexpression in a multidrug-resistant lung cancer cell line [147]. The MRP family consists of the four isoforms MRP1-4 [148, 149]. MRPs are similar to P-gp in that they are (I) capable of decreasing intracellular drug levels and (II) ATP-dependent [150]. Also, MRPs require glutathione GSH to extrude xenobiotics [151–154].
MXR
The Mitoxantrone resistance protein MXR or the Multixenobiotic resistance protein, also known as BCRP, ABCP and ABCG2, is one member of the ABC-superfamily that plays a role in trafficking biological molecules across cell membranes [155]. Expression of MXR might be an alternative strategy of resistance if cancer cells lack p-gp and MRP [156]. MXR preferentially extrudes large hydrophobic, positively charged molecules while others members of the MRP family can eject both hydrophobic uncharged molecules and water-soluble anionic compounds [157].
Alteration of drug target
When the drug reaches its target, another mechanism of resistance could be evolved somatically. Examples, which explain this mechanism of resistance, is:
-
Methotrexate is a drug of choice for the treatment of rheumatoid arthritis [158–160]. Moreover, its activity against several types of tumors has been shown. It inhibits tumor cells via inhibition of the Dihydrofolate reductase enzyme (DHFR) which is a co-enzyme in DNA-methylation. Both, in vitro and in vivo studies show that the genomic amplification of the DHFR gene is reflected by extra copies of DHFR [161–163].
-
5-fluorouracil is a thymidylate synthetase inhibitor that is widely used in several types of tumors. Thymidylate synthetase is an enzyme used to generate thymidine monophosphate, which is subsequently phosphorylated to thymidine triphosphate for use in DNA synthesis and repair [164]. It has been postulated that one mechanism of resistance is the gain of extra copies of thymidylate synthetase genes [165, 166].
Microenvironmental resistance
Cancer cells maintain a unique pH gradient; it is more acidic extracellularly and more alkaline intracellularly about normal tissues [167–169]. Such a pH gradient creates a unique environment around cancer cells. The tumor microenvironment becomes one of the cancer’s hallmarks [109]. Tumor microenvironment increases tumor’s fitness by blunting the immune system [167, 170], activating endogenous immunosuppressive strategies [171] and inhibiting the growth of the normal cell population. Moreover, the tumor microenvironment disables the activities of several chemotherapeutic agents resulting in resistance and failure in drug response [172] either through disturbing drug partitioning, sequestering it intracellularly [173, 174] or through induction of MDR expression [146].
There are several components of the tumor microenvironment that contribute to drug disability. Some of them include:
pH
Most anticancer agents are either weak bases or weak acids. Some of them are Zwitterions (see Table 2). Weakly basic anticancer drugs are ionizable at the interstitial fluid that decreases their partitioning, and if they cross the plasma membrane, they are sequestered into acidic vesicles (lysosomes). While weakly acid drugs increase their partitioning into the interstitial fluid, they will be rendered at cytosol due to intracellular alkalinity, and so they are slightly prevented from reaching their targets. This phenomenon is well known as “ion trapping mechanism” [123, 124, 175–177]. While basic drugs have reduced efficacy in an acidic microenvironment, Chlorambucil is a weakly acidic compound, and its cytotoxicity is enhanced by acidic microenvironments [178]. So, any attempt to induce intracellular acidification could be an avenue to both breaking through MDR and as an anticancer therapeutic approach it own [179].
Oxygen
Intermittent hypoxia has been considered a suggested mechanism for the initiation of carcinogenesis [180–183] tumor evolution and progression [184–186] and metastasis [187].
Hypoxia somehow handles drug resistance via the following factors:
-
1.
Most anticancer agents act to activate the apoptosis pathway, and the presence of free radicals are essential to promote this process [188]. Therefore, the absence of oxygen will diminish the activity of these drugs. Hypoxia does not only confer resistance to chemotherapy [189–192] but also to radiation [27].
-
2.
Hypoxia induces genes expression that code for ABC-transporters [193] and so favors the developing of resistance of some of the anticancer drugs e.g. 5–fluorouracil [194].
-
3.
Hypoxia also alters activity of some of metabolizing enzymes that are responsible for the activation and/or inactivation of some of the anticancer drugs e.g. conversion of paclitaxel metabolism into 6-α-hydroxypaclitaxel is reduced upon hypoxic conditions compared to normoxic conditions in HepaRG cells [195]. Therefore, hypoxia might alter therapeutic effectiveness.
-
4.
Hypoxia is an important evolutionary determinant factor that shapes part of the tumor population to become hypoxia-adapted [108]. Hypoxia is associated with cellular senescence [196]. Chemotherapeutic agents which are designed to target cells that have high proliferation rates will fail even if they reach their site of action in sufficient amounts in poorly vascularized regions [197, 198]. Conversely, recent data suggests that hypoxia suppresses geroconversion (the conversion of arrested cells to senescence) [199] which make the role of hypoxia in tumorigenesis and tumor resistance still unclear.
Glucose
In the 1920s, Otto Warburg discovered that cancer have high aerobic glycolysis even in the presence of oxygen [200–202]. Recently, metabolic reprogramming of cancer has been adopted as one of the hallmarks of cancer [109]. Glucose, especially in high loads, induces over-expression of Sodium Hydrogen Exchanger 1 (NHE-1) resulting in the alkalinization of pHi [203] and in the induction of the metabolic transformation that aggravates the tumor microenvironment [27]. Moreover, glucose uptake is associated with tumor progression [204] as well as chemotherapeutic resistance [205]. Therefore, alteration of glucose transport [206], glucose deprivation, and fasting may enhance tumor sensitivity to chemotherapy [207]. However, targeting glucose also leads to interconversion of sensitive populations into more resistant populations, which may handle relapse [208]. So, targeting glucose as a potential strategy for overcoming resistance may be a dead end.
Mesoscopic (physical, mechanical) or (regional) resistance [tumor—host interacting factors]
The physics of the tumor site has a great impact on drug activity and results in drug resistance as follows:
-
1.
Geometric, or vascular, resistance is a complex function of vascular morphology, i.e. the number of vessels of various types, their branching pattern, their diameter, and length [209]. It has been showing that upon clonal tumor expansion, tumor perfusion, and, therefore, the amount of drug to reaching its target is decreased [198, 210, 211]. Vascular modulating agents may alter vascular resistance and diminish the drugs reaching their targets [212] while by reducing the geometric resistance will enhance the activity of chemotherapeutic agents [213].
-
2.
Blood viscosity is very significant especially for intratumoral blood flow [209]. Highly blood viscosity is an indicator for blood stasis and thus for the stagnation of drugs at certain sites. Moreover, inflammatory mediators at tumor sites might alter erythrocyte sedimentation rate and lead to greatly enhanced blood viscosity [214], at least at the tumor. Because blood flow has been correlated with oxygen diffusion kinetics [215], increasing blood viscosity may induces blood flow retardation and hypoxia becomes an adaptive strategy of survival especially for xeric phenotypes (cancer cells that grow distal from blood vessels) [108]. This also contributes to drug penetration into intratumor regions (see Figure 3). Anemia also might occur due reduction in hematocrit. In this regard, bone marrow suppression by chemotherapeutic agents should be taken into consideration.
Figure 3 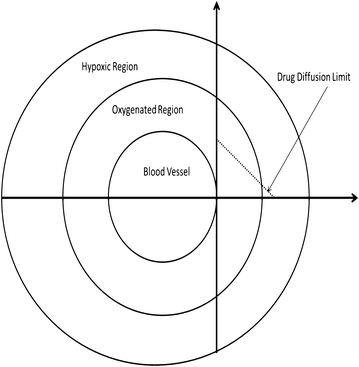
A hypothetical model describes the sphenoid tumor as mutli-habitat or multilayer shows that decrease of oxygen diffusion with drug gradients as a function of distance from the blood vessel.

Co-resistance
The presence of the extracellular matrix (ECM) and a stroma are crucial for carcinogenesis. Both of this play a significant role in cancer progression, metabolism and the metastatic cascade [216]. In this context, the ECM plays a critical role in mediating drug resistance [217], either by acting as a physical barrier that impairs drug diffusion [218] and/or by cooperating with tumor cells to generate chemotherapy resistance [219, 220]. In this regard, this strategy of resistance could be called ECM-dependent resistance [220, 221].
Conclusions
From bacteria to cancer, (multi) drug resistance is becoming a central issue and a significant challenge for medicine today. Although drug resistance is often studied at the single cell level, it is important to realize that the ability of a drug to interact with its target is more complex involving many body compartments. Also, over the past decade, there have been significant changes in our understanding of some fields from biology. Whereas, before, the key-lock model was supposed to uncover biology helping us to understand life, it is clear today that evolution theory needs to be introduced at the single cell level to clarify our understanding of some diseases including cancer. The famous key-lock model, as well as the long-awaited magic bullet to kill cancer, has to be revised accordingly. Therefore, we suggest reframing the concepts used in drug resistance in a more general context thereby dismantling the monolithic tone that the resistance is only a matter of genes. We propose that drug ineffectiveness results from tumor-host interactions and that a clear understanding of such an interaction open new opportunities not only for the discovery of new drugs but also for new therapeutic strategies to overcome the development and evolution of resistance to cancer chemotherapy.
References
Siegel R, Ma J, Zou Z, Jemal A (2014) Cancer statistics, 2014. CA Cancer J Clin 64:9–29
Gatenby RA (2009) A change of strategy in the war on cancer. Nature 459:508–509
Harguindey S, Orive G, Luis Pedraz J, Paradiso A, Reshkin SJ (2005) The role of pH dynamics and the Na+/H+ antiporter in the etiopathogenesis and treatment of cancer. Two faces of the same coin–one single nature. Biochim Biophys Acta 1756:1–24
Undevia SD, Gomez-Abuin G, Ratain MJ (2005) Pharmacokinetic variability of anticancer agents. Nat Rev Cancer 5:447–458
Paxton JW (1986) The effect of food on the bioavailability and kinetics of the anticancer drug amsacrine and a new analogue, N-5-dimethyl-9-[(2-methoxy-4-methylsulphonylamino)phenylamino]-4 acridinecarboxamide in rabbits. J Pharm Pharmacol 38:837–840
McLeod HL, Evans WE (1999) Oral cancer chemotherapy: the promise and the pitfalls. Clin Cancer Res 5:2669–2671
DeMario MD, Ratain MJ (1998) Oral chemotherapy: rationale and future directions. J Clin Oncol 16:2557–2567
Fujiwara Y (1999) Current status of oral anticancer drugs in Japan. J Clin Oncol 17:3362–3365
Sharma S (2001) Patient selection for oral chemotherapy. Oncology (Williston Park) 15(1 Suppl 2):33–35
Thörn M, Finnström N, Lundgren S, Rane A, Lööf L (2005) Cytochromes P450 and MDR1 mRNA expression along the human gastrointestinal tract. Br J Clin Pharmacol 60:54–60
Wacher VJ, Salphati L, Benet LZ (2001) Active secretion and enterocytic drug metabolism barriers to drug absorption. Adv Drug Deliv Rev 46:89–102
Sparreboom A, van Asperen J, Mayer U, Schinkel AH, Smit JW, Meijer DKF et al (1997) Limited oral bioavailability and active epithelial excretion of paclitaxel (Taxol) caused by P-glycoprotein in the intestine. Proc Natl Acad Sci USA 94:2031–2035
Burger H, Nooter K (2004) Pharmacokinetic resistance to imatinib mesylate: role of the ABC drug pumps ABCG2 (BCRP) and ABCB1 (MDR1) in the oral bioavailability of imatinib. Cell Cycle 3:1502–1505
Dietrich CG, Geier A, Oude Elferink RPJ (2003) ABC of oral bioavailability: transporters as gatekeepers in the gut. Gut 52:1788–1795
Cascorbi I, Gerloff T, Johne A, Meisel C, Hoffmeyer S, Schwab M et al (2001) Frequency of single nucleotide polymorphisms in the P-glycoprotein drug transporter MDR1 gene in white subjects. Clin Pharmacol Ther 69:169–174
Ling J, Fettner S, Lum BL, Riek M, Rakhit A (2008) Effect of food on the pharmacokinetics of erlotinib, an orally active epidermal growth factor receptor tyrosine-kinase inhibitor, in healthy individuals. Anticancer Drugs 19:209–216
Jain RK, Brar SS, Lesko LJ (2010) Food and oral antineoplastics: more than meets the eye. Clin Cancer Res 16:4305–4307
Kang SP, Ratain MJ (2010) Inconsistent labeling of food effect for oral agents across therapeutic areas: differences between oncology and non-oncology products. Clin Cancer Res 16:4446–4451
Herben VM, Rosing H, ten Bokkel Huinink WW, van Zomeren DM, Batchelor D, Doyle E (1999) Oral topotecan: bioavailablity and effect of food co-administration. Br J Cancer 80:1380–1386
Kivistö KT, Niemi M, Fromm MF (2004) Functional interaction of intestinal CYP3A4 and P-glycoprotein. Fundam Clin Pharmacol 18:621–626
He K, Iyer KR, Hayes RN, Sinz MW, Woolf TF, Hollenberg PF (1998) Inactivation of cytochrome P450 3A4 by bergamottin, a component of grapefruit juice. Chem Res Toxicol 11:252–259
Fujita K (2004) Food-drug interactions via human cytochrome P450 3A (CYP3A). Drug Metabol Drug Interact 20:195–217
Singh BN, Malhotra BK (2004) Effects of food on the clinical pharmacokinetics of anticancer agents: underlying mechanisms and implications for oral chemotherapy. Clin Pharmacokinet 43:1127–1156
Ruggiero A, Cefalo MG, Coccia P, Mastrangelo S, Maurizi P, Riccardi R (2012) The role of diet on the clinical pharmacology of oral antineoplastic agents. Eur J Clin Pharmacol 68:115–122
Wilson K (1984) Sex-related differences in drug disposition in man. Clin Pharmacokinet 9:189–202
Pleym H, Spigset O, Kharasch ED, Dale O (2003) Gender differences in drug effects: implications for anesthesiologists. Acta Anaesthesiol Scand 47:241–259
Alfarouk KO, Shayoub MEAA, Muddathir AK, Elhassan GO, Bashir AHHH (2011) Evolution of tumor metabolism might reflect carcinogenesis as a reverse evolution process (dismantling of multicellularity). Cancers (Basel) 3:3002–3017
Soldin OP, Chung SH, Mattison DR (2011) Sex differences in drug disposition. J Biomed Biotechnol 2011:187103
Beierle I, Meibohm B, Derendorf H (1999) Gender differences in pharmacokinetics and pharmacodynamics. Int J Clin Pharmacol Ther 37:529–547
Hanley MJ, Abernethy DR, Greenblatt DJ (2010) Effect of obesity on the pharmacokinetics of drugs in humans. Clin Pharmacokinet 49:71–87
Theologides A (1977) Nutritional management of the patient with advanced cancer. Postgrad Med 61:97–101
Fearon KC (1992) The Sir David Cuthbertson Medal Lecture 1991. The mechanisms and treatment of weight loss in cancer. Proc Nutr Soc 51:251–265
Fearon KC, Voss AC, Hustead DS (2006) Definition of cancer cachexia: effect of weight loss, reduced food intake, and systemic inflammation on functional status and prognosis. Am J Clin Nutr 83:1345–1350
Powis G, Reece P, Ahmann DL, Ingle JN (1987) Effect of body weight on the pharmacokinetics of cyclophosphamide in breast cancer patients. Cancer Chemother Pharmacol 20:219–222
Felici A, Verweij J, Sparreboom A (2002) Dosing strategies for anticancer drugs: the good, the bad and body-surface area. Eur J Cancer 38:1677–1684
Tillement JP, Zini R, d’ Athis P, Vassent G (1974) Binding of certain acidic drugs to human albumin: theoretical and practical estimation of fundamental parameters. Eur J Clin Pharmacol 7:307–313
Ruiz-Cabello F, Erill S (1984) Abnormal serum protein binding of acidic drugs in diabetes mellitus. Clin Pharmacol Ther 36:691–695
Routledge PA (1986) The plasma protein binding of basic drugs. Br J Clin Pharmacol 22:499–506
Goldwasser P, Feldman J (1997) Association of serum albumin and mortality risk. J Clin Epidemiol 50:693–703
Lis CG, Grutsch JF, Vashi PG, Lammersfeld CA (2003) Is serum albumin an independent predictor of survival in patients with breast cancer? JPEN J Parenter Enteral Nutr 27:10–15
Grimm G, Haslacher H, Kampitsch T, Endler G, Marsik C, Schickbauer T et al (2009) Sex differences in the association between albumin and all-cause and vascular mortality. Eur J Clin Invest 39:860–865
Liu B, Earl HM, Poole CJ, Dunn J, Kerr DJ (1995) Etoposide protein binding in cancer patients. Cancer Chemother Pharmacol 36:506–512
Liliemark E, Söderhäll S, Sirzea F, Gruber A, Osby E, Björkholm M et al (1996) Higher in vivo protein binding of etoposide in children compared with adult cancer patients. Cancer Lett 106:97–100
Gambacorti-Passerini C, Zucchetti M, Russo D, Frapolli R, Verga M, Bungaro S et al (2003) Alpha1 acid glycoprotein binds to imatinib (STI571) and substantially alters its pharmacokinetics in chronic myeloid leukemia patients. Clin Cancer Res 9:625–632
Li J, Brahmer J, Messersmith W, Hidalgo M, Baker SD (2006) Binding of gefitinib, an inhibitor of epidermal growth factor receptor-tyrosine kinase, to plasma proteins and blood cells: in vitro and in cancer patients. Invest New Drugs 24:291–297
Wu J, Lorusso PM, Matherly LH, Li J (2012) Implications of plasma protein binding for pharmacokinetics and pharmacodynamics of the γ-secretase inhibitor RO4929097. Clin Cancer Res 18:2066–2079
Levi F, Schibler U (2007) Circadian rhythms: mechanisms and therapeutic implications. Annu Rev Pharmacol Toxicol 47:593–628
Sukumaran S, Almon RR, DuBois DC, Jusko WJ (2010) Circadian rhythms in gene expression: relationship to physiology, disease, drug disposition and drug action. Adv Drug Deliv Rev 62:904–917
Takahashi JS, Hong H-K, Ko CH, McDearmon EL (2008) The genetics of mammalian circadian order and disorder: implications for physiology and disease. Nat Rev Genet 9:764–775
Rana S, Mahmood S (2010) Circadian rhythm and its role in malignancy. J Circadian Rhythms 8:3
Straub RH, Cutolo M (2007) Circadian rhythms in rheumatoid arthritis: implications for pathophysiology and therapeutic management. Arthritis Rheum 56:399–408
Lévi F, Okyar A, Dulong S, Innominato PF, Clairambault J (2010) Circadian timing in cancer treatments. Annu Rev Pharmacol Toxicol 50:377–421
Prémaud A, Rousseau A, Gicquel M, Ragot S, Manceau J, Laurentie M et al (2002) An animal model for the study of chronopharmacokinetics of drugs and application to methotrexate and vinorelbine. Toxicol Appl Pharmacol 183:189–197
Baraldo M (2008) The influence of circadian rhythms on the kinetics of drugs in humans. Expert Opin Drug Metab Toxicol 4:175–192
Erkekoglu P, Baydar T (2012) Chronopharmacokinetics of drugs in toxicological aspects: a short review for pharmacy practitioners. J Res Pharm Pract 1:3–9
Touitou Y, Touitou C, Bogdan A, Reinberg A, Auzeby A, Beck H et al (1986) Differences between young and elderly subjects in seasonal and circadian variations of total plasma proteins and blood volume as reflected by hemoglobin, hematocrit, and erythrocyte counts. Clin Chem 32:801–804
Bruguerolle B, Arnaud C, Levi F, Focan C, Touitou Y, Bouvenot G (1989) Physiopathological alterations of alpha 1 acid glycoprotein temporal variations: implications for chronopharmacology. Prog Clin Biol Res 300:199–214
Hecquet B, Meynadier J, Bonneterre J, Adenis L, Demaille A (1985) Time dependency in plasmatic protein binding of cisplatin. Cancer Treat Rep 69:79–83
Hecquet B, Sucche M (1986) Theoretical study of the influence of the circadian rhythm of plasma protein binding on cisplatin area under the curve. J Pharmacokinet Biopharm 14:79–93
Lévi F (2006) Chronotherapeutics: the relevance of timing in cancer therapy. Cancer Causes Control 17:611–621
Guengerich FP (2007) Mechanisms of cytochrome P450 substrate oxidation: MiniReview. J Biochem Mol Toxicol 21:163–168
Guengerich FP (2008) Cytochrome p450 and chemical toxicology. Chem Res Toxicol 21:70–83
Jancova P, Anzenbacher P, Anzenbacherova E (2010) Phase II drug metabolizing enzymes. Biomed Pap Med Fac Univ Palacky Olomouc Czech Repub 154:103–116
Gonzalez FJ (2006) Goodman and Gilman’s Pharmacological Basis of Therapeutics. In: Brunton LLLL, Lazo JS (eds) The McGraw-Hill Companies, Inc, New York, pp 71–92
Rooseboom M, Commandeur JNM, Vermeulen NPE (2004) Enzyme-catalyzed activation of anticancer prodrugs. Pharmacol Rev 56:53–102
Akhdar H, Legendre C, Caroline Aninat FM (2012) Topics on drug metabolism. In: Paxton J (ed) INTECH, pp 137–170
McFadyen MCE, Melvin WT, Murray GI (2004) Cytochrome P450 enzymes: novel options for cancer therapeutics. Mol Cancer Ther 3:363–371
Rodriguez-Antona C, Ingelman-Sundberg M (2006) Cytochrome P450 pharmacogenetics and cancer. Oncogene 25:1679–1691
Lin JH, Lu AY (2001) Interindividual variability in inhibition and induction of cytochrome P450 enzymes. Annu Rev Pharmacol Toxicol 41:535–567
Ingelman-Sundberg M, Sim SC, Gomez A, Rodriguez-Antona C (2007) Influence of cytochrome P450 polymorphisms on drug therapies: pharmacogenetic, pharmacoepigenetic and clinical aspects. Pharmacol Ther 116:496–526
Plant N (2007) The human cytochrome P450 sub-family: transcriptional regulation, inter-individual variation and interaction networks. Biochim Biophys Acta 1770:478–488
Hayes JD, Pulford DJ (1995) The glutathione S-transferase supergene family: regulation of GST and the contribution of the isoenzymes to cancer chemoprotection and drug resistance. Crit Rev Biochem Mol Biol 30:445–600
Xu C, Li CY-T, Kong A-NT (2005) Induction of phase I, II and III drug metabolism/transport by xenobiotics. Arch Pharm Res 28:249–268
Nakagawa K, Saijo N, Tsuchida S, Sakai M, Tsunokawa Y, Yokota J et al (1990) Glutathione-S-transferase pi as a determinant of drug resistance in transfectant cell lines. J Biol Chem 265:4296–4301
Uozaki H, Horiuchi H, Ishida T, Iijima T, Imamura T, Machinami R (1997) Overexpression of resistance-related proteins (metallothioneins, glutathione-S-transferase pi, heat shock protein 27, and lung resistance-related protein) in osteosarcoma. Relationship with poor prognosis. Cancer 79:2336–2344
Harkey MA, Czerwinski M, Slattery J, Kiem H-P (2005) Overexpression of glutathione-S-transferase, MGSTII, confers resistance to busulfan and melphalan. Cancer Invest 23:19–25
Kauvar LM, Morgan AS, Sanderson PE, Henner WD (1998) Glutathione based approaches to improving cancer treatment. Chem Biol Interact 111–112:225–238
Riddick DS, Lee C, Ramji S, Chinje EC, Cowen RL, Williams KJ et al (2005) Cancer chemotherapy and drug metabolism. Drug Metab Dispos 33:1083–1096
Kodym R, Calkins P, Story M (1999) The cloning and characterization of a new stress response protein. A mammalian member of a family of theta class glutathione s-transferase-like proteins. J Biol Chem 274:5131–5137
Cumming RC, Lightfoot J, Beard K, Youssoufian H, O’Brien PJ, Buchwald M (2001) Fanconi anemia group C protein prevents apoptosis in hematopoietic cells through redox regulation of GSTP1. Nat Med 7:814–820
Townsend DM, Tew KD (2003) The role of glutathione-S-transferase in anti-cancer drug resistance. Oncogene 22:7369–7375
Gunnarsdottir S, Elfarra AA (1999) Glutathione-dependent metabolism of cis-3-(9H-purin-6-ylthio)acrylic acid to yield the chemotherapeutic drug 6-mercaptopurine: evidence for two distinct mechanisms in rats. J Pharmacol Exp Ther 290:950–957
Tew KD (2005) TLK-286: a novel glutathione S-transferase-activated prodrug. Expert Opin Investig Drugs 14:1047–1054
Nelson HH, Wiencke JK, Christiani DC, Cheng TJ, Zuo ZF, Schwartz BS et al (1995) Ethnic differences in the prevalence of the homozygous deleted genotype of glutathione S-transferase theta. Carcinogenesis 16:1243–1245
Kempkes M, Golka K, Reich S, Reckwitz T, Bolt HM (1996) Glutathione S-transferase GSTM1 and GSTT1 null genotypes as potential risk factors for urothelial cancer of the bladder. Arch Toxicol 71:123–126
LA Magno V, Talbot J, Talbot T, Borges Santos AM, Souza RP, Marin LJ (2009) Glutathione s-transferase variants in a brazilian population. Pharmacology 83:231–236
Ying X-J, Dong P, Shen B, Xu C-Z, Xu H-M, Zhao S-W (2012) Glutathione S-transferase M1 gene polymorphism and laryngeal cancer risk: a meta-analysis. PLoS One 7:e42826
Liu J, Chen H, Miller DS, Saavedra JE, Keefer LK, Johnson DR et al (2001) Overexpression of glutathione S-transferase II and multidrug resistance transport proteins is associated with acquired tolerance to inorganic arsenic. Mol Pharmacol 60:302–309
Lévi F, Altinok A, Clairambault J, Goldbeter A (2008) Implications of circadian clocks for the rhythmic delivery of cancer therapeutics. Philos Trans A Math Phys Eng Sci 366:3575–3598
Zeng Z-L, Sun J, Guo L, Li S, Wu M, Qiu F et al (2005) Circadian rhythm in dihydropyrimidine dehydrogenase activity and reduced glutathione content in peripheral blood of nasopharyngeal carcinoma patients. Chronobiol Int 22:741–754
Smaaland R, Sothern RB, Laerum OD, Abrahamsen JF (2002) Rhythms in human bone marrow and blood cells. Chronobiol Int 19:101–127
Krishna DR, Klotz U (1994) Extrahepatic metabolism of drugs in humans. Clin Pharmacokinet 26:144–160
Pacifici GM, Franchi M, Bencini C, Repetti F, Di Lascio N, Muraro GB (1988) Tissue distribution of drug-metabolizing enzymes in humans. Xenobiotica 18:849–856
Fisher MB, Paine MF, Strelevitz TJ, Wrighton SA (2001) The role of hepatic and extrahepatic UDP-glucuronosyltransferases in human drug metabolism. Drug Metab Rev 33:273–297
Ritter JK (2007) Intestinal UGTs as potential modifiers of pharmacokinetics and biological responses to drugs and xenobiotics. Expert Opin Drug Metab Toxicol 3:93–107
Dai D, Zeldin DC, Blaisdell JA, Chanas B, Coulter SJ, Ghanayem BI et al (2001) Polymorphisms in human CYP2C8 decrease metabolism of the anticancer drug paclitaxel and arachidonic acid. Pharmacogenetics 11:597–607
Gréen H, Söderkvist P, Rosenberg P, Mirghani RA, Rymark P, Lundqvist EA et al (2009) Pharmacogenetic studies of Paclitaxel in the treatment of ovarian cancer. Basic Clin Pharmacol Toxicol 104:130–137
Kusuhara H, Sugiyama Y (2002) Role of transporters in the tissue-selective distribution and elimination of drugs: transporters in the liver, small intestine, brain and kidney. J Control Release 78:43–54
Vaisman BL, Lambert G, Amar M, Joyce C, Ito T, Shamburek RD et al (2001) ABCA1 overexpression leads to hyperalphalipoproteinemia and increased biliary cholesterol excretion in transgenic mice. J Clin Invest 108:303–309
Yu L, Li-Hawkins J, Hammer RE, Berge KE, Horton JD, Cohen JC et al (2002) Overexpression of ABCG5 and ABCG8 promotes biliary cholesterol secretion and reduces fractional absorption of dietary cholesterol. J Clin Invest 110:671–680
Vlaming MLH, Mohrmann K, Wagenaar E, de Waart DR, Elferink RPJO, Lagas JS et al (2006) Carcinogen and anticancer drug transport by Mrp2 in vivo: studies using Mrp2 (Abcc2) knockout mice. J Pharmacol Exp Ther 318:319–327
Figge A, Lammert F, Paigen B, Henkel A, Matern S, Korstanje R et al (2004) Hepatic overexpression of murine Abcb11 increases hepatobiliary lipid secretion and reduces hepatic steatosis. J Biol Chem 279:2790–2799
Vree TB, Van Ewijk-Beneken Kolmer EW, Wuis EW, Hekster YA, Broekman MM (1993) Interindividual variation in the capacity-limited renal glucuronidation of probenecid by humans. Pharm World Sci 15:197–202
Morris ME, Lee H-J, Predko LM (2003) Gender differences in the membrane transport of endogenous and exogenous compounds. Pharmacol Rev 55:229–240
Johnson JA (2000) Predictability of the effects of race or ethnicity on pharmacokinetics of drugs. Int J Clin Pharmacol Ther 38:53–60
Loadman PM, Bibby MC (1994) Pharmacokinetic drug interactions with anticancer drugs. Clin Pharmacokinet 26:486–500
Siemann DW (2006) Tumor vasculature : a target for anticancer therapies. In: Vascular-targeted therapies in oncology. John Wiley & Sons, Ltd, Chichester, pp 1–8
Alfarouk KO, Ibrahim ME, Gatenby RA, Brown JS (2013) Riparian ecosystems in human cancers. Evol Appl 6:46–53
Hanahan D, Weinberg RA (2011) Hallmarks of cancer: the next generation. Cell 144:646–674
Ma J, Waxman DJ (2008) Combination of antiangiogenesis with chemotherapy for more effective cancer treatment. Mol Cancer Ther 7:3670–3684
Kerbel RS (2006) Antiangiogenic therapy: a universal chemosensitization strategy for cancer? Science 312:1171–1175
Lloyd MC, Alfarouk KO, Verduzco D, Bui MM, Gillies RJ, Ibrahim ME et al (2014) Vascular measurements correlate with estrogen receptor status. BMC Cancer 14:279
Robey IF, Baggett BK, Kirkpatrick ND, Roe DJ, Dosescu J, Sloane BF et al (2009) Bicarbonate increases tumor pH and inhibits spontaneous metastases. Cancer Res 69:2260–2268
Lippens RJ (1984) Methotrexate. I. Pharmacology and pharmacokinetics. Am J Pediatr Hematol Oncol 6:379–395
Singh MS, Francis PA, Michael M (2011) Tamoxifen, cytochrome P450 genes and breast cancer clinical outcomes. Breast 20:111–118
Stearns V, Johnson MD, Rae JM, Morocho A, Novielli A, Bhargava P et al (2003) Active tamoxifen metabolite plasma concentrations after coadministration of tamoxifen and the selective serotonin reuptake inhibitor paroxetine. J Natl Cancer Inst 95:1758–1764
Caraci F, Crupi R, Drago F, Spina E (2011) Metabolic drug interactions between antidepressants and anticancer drugs: focus on selective serotonin reuptake inhibitors and hypericum extract. Curr Drug Metab 12:570–577
Yap KYL, Tay WL, Chui WK, Chan A (2011) Clinically relevant drug interactions between anticancer drugs and psychotropic agents. Eur J Cancer Care (Engl) 20:6–32
Yamazaki M, Suzuki H, Sugiyama Y (1996) Recent advances in carrier-mediated hepatic uptake and biliary excretion of xenobiotics. Pharm Res 13:497–513
König J, Nies AT, Cui Y, Leier I, Keppler D (1999) Conjugate export pumps of the multidrug resistance protein (MRP) family: localization, substrate specificity, and MRP2-mediated drug resistance. Biochim Biophys Acta 1461:377–394
Hirano M, Maeda K, Hayashi H, Kusuhara H, Sugiyama Y (2005) Bile salt export pump (BSEP/ABCB11) can transport a nonbile acid substrate, pravastatin. J Pharmacol Exp Ther 314:876–882
Kamisako T, Ogawa H (2004) Effects of pravastatin and bezafibrate on biliary lipid excretion and hepatic expression of Abcg5 and Abcg8 in the rat. J Gastroenterol Hepatol 19:879–883
Raghunand N, Martínez-Zaguilán R, Wright SH, Gillies RJ (1999) pH and drug resistance. II. Turnover of acidic vesicles and resistance to weakly basic chemotherapeutic drugs. Biochem Pharmacol 57:1047–1058
Mahoney BP, Raghunand N, Baggett B, Gillies RJ (2003) Tumor acidity, ion trapping and chemotherapeutics. I. Acid pH affects the distribution of chemotherapeutic agents in vitro. Biochem Pharmacol 66:1207–1218
Gottesman MM (2002) Mechanisms of cancer drug resistance. Annu Rev Med 53:615–627
Stewart DJ (2007) Saturable passive resistance in chemotherapy of epithelial maligancies. In: Parsons RA (ed) Progress in cancer drug resistance research. Nova Science, New York, pp 29–41
De Visser KE, Jonkers J (2009) Towards understanding the role of cancer-associated inflammation in chemoresistance. Curr Pharm Des 15:1844–1853
Barlow M, Hall BG (2002) Phylogenetic analysis shows that the OXA β-lactamase genes have been on plasmids for millions of years. J Mol Evol 55:314–321
Hall BG, Barlow M (2004) Evolution of the serine beta-lactamases: past, present and future. Drug Resist Updat 7:111–123
Baltz RH (2008) Renaissance in antibacterial discovery from actinomycetes. Curr Opin Pharmacol 557–563
D’Costa VM, King CE, Kalan L, Morar M, Sung WWL, Schwarz C et al (2011) Antibiotic resistance is ancient. Nature 477:457–461
Bhullar K, Waglechner N, Pawlowski A, Koteva K, Banks ED, Johnston MD et al (2012) Antibiotic resistance is prevalent in an isolated cave microbiome. PLoS One 7:e34953
Ueda K, Clark DP, Chen CJ, Roninson IB, Gottesman MM, Pastan I (1987) The human multidrug resistance (mdr1) gene. cDNA cloning and transcription initiation. J Biol Chem 262:505–508
Goldstein LJ, Galski H, Fojo A, Willingham M, Lai SL, Gazdar A et al (1989) Expression of a multidrug resistance gene in human cancers. J Natl Cancer Inst 81:116–124
Zhou S-F (2008) Structure, function and regulation of P-glycoprotein and its clinical relevance in drug disposition. Xenobiotica 38:802–832
Thiebaut F, Tsuruo T, Hamada H, Gottesman MM, Pastan I, Willingham MC (1987) Cellular localization of the multidrug-resistance gene product P-glycoprotein in normal human tissues. Proc Natl Acad Sci USA 84:7735–7738
Schinkel A (1999) P-Glycoprotein, a gatekeeper in the blood-brain barrier. Adv Drug Deliv Rev 36:179–194
Maliepaard M, Scheffer GL, Faneyte IF, van Gastelen MA, Pijnenborg AC, Schinkel AH et al (2001) Subcellular localization and distribution of the breast cancer resistance protein transporter in normal human tissues. Cancer Res 61:3458–3464
Schuetz EG, Schinkel AH, Relling MV, Schuetz JD (1996) P-glycoprotein: a major determinant of rifampicin-inducible expression of cytochrome P4503A in mice and humans. Proc Natl Acad Sci USA 93:4001–4005
Munteanu E, Verdier M, Grandjean-Forestier F, Stenger C, Jayat-Vignoles C, Huet S et al (2006) Mitochondrial localization and activity of P-glycoprotein in doxorubicin-resistant K562 cells. Biochem Pharmacol 71:1162–1174
Bendayan R, Ronaldson PT, Gingras D, Bendayan M (2006) In situ localization of P-glycoprotein (ABCB1) in human and rat brain. J Histochem Cytochem 54:1159–1167
Solazzo M, Fantappiè O, D’Amico M, Sassoli C, Tani A, Cipriani G et al (2009) Mitochondrial expression and functional activity of breast cancer resistance protein in different multiple drug-resistant cell lines. Cancer Res 69:7235–7242
Shen Y, Chu Y, Yang Y, Wang Z (2012) Mitochondrial localization of P-glycoprotein in the human breast cancer cell line MCF-7/ADM and its functional characterization. Oncol Rep 27:1535–1540
Solazzo M, Fantappiè O, Lasagna N, Sassoli C, Nosi D, Mazzanti R (2006) P-gp localization in mitochondria and its functional characterization in multiple drug-resistant cell lines. Exp Cell Res 312:4070–4078
Babakhanian K, Bendayan M, Bendayan R (2007) Localization of P-glycoprotein at the nuclear envelope of rat brain cells. Biochem Biophys Res Commun 361:301–306
Wei LY, Roepe PD (1994) Low external pH and osmotic shock increase the expression of human MDR protein. Biochemistry 33:7229–7238
Cole SP, Bhardwaj G, Gerlach JH, Mackie JE, Grant CE, Almquist KC et al (1992) Overexpression of a transporter gene in a multidrug-resistant human lung cancer cell line. Science 258:1650–1654
Young LC, Campling BG, Cole SP, Deeley RG, Gerlach JH (2001) Multidrug resistance proteins MRP3, MRP1, and MRP2 in lung cancer: correlation of protein levels with drug response and messenger RNA levels. Clin Cancer Res 7:1798–1804
Copsel S, Garcia C, Diez F, Vermeulem M, Baldi A, Bianciotti LG et al (2011) Multidrug resistance protein 4 (MRP4/ABCC4) regulates cAMP cellular levels and controls human leukemia cell proliferation and differentiation. J Biol Chem 286:6979–6988
Sauna ZE, Ambudkar SV (2007) About a switch: how P-glycoprotein (ABCB1) harnesses the energy of ATP binding and hydrolysis to do mechanical work. Mol Cancer Ther 6:13–23
Keppler D, Leier I, Jedlitschky G, König J (1998) ATP-dependent transport of glutathione S-conjugates by the multidrug resistance protein MRP1 and its apical isoform MRP2. Chem Biol Interact 111–112:153–161
Keppler D (1999) Export pumps for glutathione S-conjugates. Free Radic Biol Med 27:985–991
Coley HM (2008) Mechanisms and strategies to overcome chemotherapy resistance in metastatic breast cancer. Cancer Treat Rev 34:378–390
Cole SPC, Deeley RG (2006) Transport of glutathione and glutathione conjugates by MRP1. Trends Pharmacol Sci 27:438–446
Litman T, Druley TE, Stein WD, Bates SE (2001) From MDR to MXR: new understanding of multidrug resistance systems, their properties and clinical significance. Cell Mol Life Sci 58:931–959
Sauna ZE, Smith MM, Müller M, Kerr KM, Ambudkar SV (2001) The mechanism of action of multidrug-resistance-linked P-glycoprotein. J Bioenerg Biomembr 33:481–491
Glavinas H, Krajcsi P, Cserepes J, Sarkadi B (2004) The role of ABC transporters in drug resistance, metabolism and toxicity. Curr Drug Deliv 1:27–42
Maini R, St Clair EW, Breedveld F, Furst D, Kalden J, Weisman M (1999) Infliximab (chimeric anti-tumour necrosis factor alpha monoclonal antibody) versus placebo in rheumatoid arthritis patients receiving concomitant methotrexate: a randomised phase III trial. ATTRACT Study Group. Lancet 354:1932–1939
Cress RH, Deaver NL (1964) Methotrexate in the management of severe psoriasis and arthritis: report of a case. South Med J 57:1088–1090
Dale J, Alcorn N, Capell H, Madhok R (2007) Combination therapy for rheumatoid arthritis: methotrexate and sulfasalazine together or with other DMARDs. Nat Clin Pract Rheumatol 3:450–458 quiz, following 478
Curt GA, Carney DN, Cowan KH, Jolivet J, Bailey BD, Drake JC et al (1983) Unstable methotrexate resistance in human small-cell carcinoma associated with double minute chromosomes. N Engl J Med 308:199–202
Schimke RT (1984) Gene amplification, drug resistance, and cancer. Cancer Res 44:1735–1742
Kobayashi S, Boggon TJ, Dayaram T, Jänne PA, Kocher O, Meyerson M et al (2005) EGFR mutation and resistance of non-small-cell lung cancer to gefitinib. N Engl J Med 352:786–792
Longley DB, Harkin DP, Johnston PG (2003) 5-fluorouracil: mechanisms of action and clinical strategies. Nat Rev Cancer 3:330–338
Watson RG, Muhale F, Thorne LB, Yu J, O’Neil BH, Hoskins JM et al (2010) Amplification of thymidylate synthetase in metastatic colorectal cancer patients pretreated with 5-fluorouracil-based chemotherapy. Eur J Cancer 46:3358–3364
Wang T-L, Diaz LA, Romans K, Bardelli A, Saha S, Galizia G et al (2004) Digital karyotyping identifies thymidylate synthase amplification as a mechanism of resistance to 5-fluorouracil in metastatic colorectal cancer patients. Proc Natl Acad Sci USA 101:3089–3094
Reshkin SJ, Cardone RA, Harguindey S (2013) Na+-H+ exchanger, pH regulation and cancer. Recent Pat Anticancer Drug Discov 8:85–99
Neri D, Supuran CT (2011) Interfering with pH regulation in tumours as a therapeutic strategy. Nat Rev Drug Discov 10:767–777
Huber V, De Milito A, Harguindey S, Reshkin SJ, Wahl ML, Rauch C et al (2010) Proton dynamics in cancer. J Transl Med 8:57
Alfarouk KO, Muddathir AK, Shayoub MEA (2011) Tumor acidity as evolutionary spite. Cancers (Basel) 3:408–414
Hao N-B, Lü M-H, Fan Y-H, Cao Y-L, Zhang Z-R, Yang S-M (2012) Macrophages in tumor microenvironments and the progression of tumors. Clin Dev Immunol 2012:948098
Correia AL, Bissell MJ (2012) The tumor microenvironment is a dominant force in multidrug resistance. Drug Resist Updat 15:39–49
Gillies RJ, Raghunand N, Karczmar GS, Bhujwalla ZM (2002) MRI of the tumor microenvironment. J Magn Reson Imaging 16:430–450
Raghunand N, Gillies RJ (2000) pH and drug resistance in tumors. Drug Resist Updat 3:39–47
Wojtkowiak JW, Verduzco D, Schramm KJ, Gillies RJ (2011) Drug resistance and cellular adaptation to tumor acidic pH microenvironment. Mol Pharm 8:2032–2038
Raghunand N, Mahoney BP, Gillies RJ (2003) Tumor acidity, ion trapping and chemotherapeutics. II. pH-dependent partition coefficients predict importance of ion trapping on pharmacokinetics of weakly basic chemotherapeutic agents. Biochem Pharmacol 66:1219–1229
Gong Y, Duvvuri M, Krise JP (2003) Separate roles for the Golgi apparatus and lysosomes in the sequestration of drugs in the multidrug-resistant human leukemic cell line HL-60. J Biol Chem 278:50234–50239
Parkins CS, Chadwick JA, Chaplin DJ (1996) Inhibition of intracellular pH control and relationship to cytotoxicity of chlorambucil and vinblastine. Br J Cancer Suppl 27:S75–S77
Alfarouk KO, Verduzco D, Rauch C, Muddathir AK, Adil HHB et al (2014) Glycolysis, tumor metabolism, cancer growth and dissemination. A new pH-based etiopathogenic perspective and therapeutic approach to an old cancer question. Oncoscience 1:777–802
Harguindey S, Pedraz JL, García Cañero R, Pérez de Diego J, Cragoe EJ (1995) Hydrogen ion-dependent oncogenesis and parallel new avenues to cancer prevention and treatment using a H(+)-mediated unifying approach: pH-related and pH-unrelated mechanisms. Crit Rev Oncog 6:1–33
Harguindey S, Arranz JL, Polo Orozco JD, Rauch C, Fais S, Cardone RA et al (2013) Cariporide and other new and powerful NHE1 inhibitors as potentially selective anticancer drugs—an integral molecular/biochemical/metabolic/clinical approach after 100 years of cancer research. J Transl Med 11:282
Van Meir E (1996) Hypoxia-mediated selection of cells with diminished apoptotic potential to solid tumours. Neurosurgery 39:878–879
Gatenby RA, Smallbone K, Maini PK, Rose F, Averill J, Nagle RB et al (2007) Cellular adaptations to hypoxia and acidosis during somatic evolution of breast cancer. Br J Cancer 97:646–653
Graeber TG, Osmanian C, Jacks T, Housman DE, Koch CJ, Lowe SW et al (1996) Hypoxia-mediated selection of cells with diminished apoptotic potential in solid tumours. Nature 379:88–91
Coquelle A, Toledo F, Stern S, Bieth A, Debatisse M (1998) A new role for hypoxia in tumor progression: induction of fragile site triggering genomic rearrangements and formation of complex DMs and HSRs. Mol Cell 2:259–265
Vaupel P (2004) The role of hypoxia-induced factors in tumor progression. Oncologist 9(Suppl 5):10–17
Bertout JA, Patel SA, Simon MC (2008) The impact of O2 availability on human cancer. Nat Rev Cancer 8:967–975
Sakata K, Kwok TT, Murphy BJ, Laderoute KR, Gordon GR, Sutherland RM (1991) Hypoxia-induced drug resistance: comparison to P-glycoprotein-associated drug resistance. Br J Cancer 64:809–814
Mattern J, Volm M (1998) Role of oxygenation and vascularization in drug resistance. Cytotechnology 27:249–256
Suzuki Y, Tanaka K, Neghishi D, Shimizu M, Murayama N, Hashimoto T et al (2008) Increased distribution of carboplatin, an anti-cancer agent, to rat brains with the aid of hyperbaric oxygenation. Xenobiotica 38:1471–1475
Lara PC, Lloret M, Clavo B, Apolinario RM, Henríquez-Hernández LA, Bordón E et al (2009) Severe hypoxia induces chemo-resistance in clinical cervical tumors through MVP over-expression. Radiat Oncol 4:29
Kim JW, Ho WJ, Wu BM (2011) The role of the 3D environment in hypoxia-induced drug and apoptosis resistance. Anticancer Res 31:3237–3245
Cosse J-P, Michiels C (2008) Tumour hypoxia affects the responsiveness of cancer cells to chemotherapy and promotes cancer progression. Anticancer Agents Med Chem 8:790–797
Zhu H, Chen XP, Luo SF, Guan J, Zhang WG, Zhang BX (2005) Involvement of hypoxia-inducible factor-1-alpha in multidrug resistance induced by hypoxia in HepG2 cells. J Exp Clin Cancer Res 24:565–574
Legendre C, Hori T, Loyer P, Aninat C, Ishida S, Glaise D et al (2009) Drug-metabolising enzymes are down-regulated by hypoxia in differentiated human hepatoma HepaRG cells: HIF-1alpha involvement in CYP3A4 repression. Eur J Cancer 45:2882–2892
Sullivan R, Paré GC, Frederiksen LJ, Semenza GL, Graham CH (2008) Hypoxia-induced resistance to anticancer drugs is associated with decreased senescence and requires hypoxia-inducible factor-1 activity. Mol Cancer Ther 7:1961–1973
Primeau AJ, Rendon A, Hedley D, Lilge L, Tannock IF (2005) The distribution of the anticancer drug Doxorubicin in relation to blood vessels in solid tumors. Clin Cancer Res 11(24 Pt 1):8782–8788
Minchinton AI, Tannock IF (2006) Drug penetration in solid tumours. Nat Rev Cancer 6:583–592
Leontieva OV, Natarajan V, Demidenko ZN, Burdelya LG, Gudkov AV, Blagosklonny MV (2012) Hypoxia suppresses conversion from proliferative arrest to cellular senescence. Proc Natl Acad Sci USA 109:13314–13318
Warburg O (1956) On the origin of cancer cells. Science 123:309–314
Weinhouse S (1956) On respiratory impairment in cancer cells. Science 124:267–269
Gatenby RA, Gillies RJ (2004) Why do cancers have high aerobic glycolysis? Nat Rev Cancer 4:891–899
Williams B, Howard RL (1994) Glucose-induced changes in Na+/H+ antiport activity and gene expression in cultured vascular smooth muscle cells. Role of protein kinase C. J Clin Invest 93:2623–2631
Fu Y, Lee AS (2006) Glucose regulated proteins in cancer progression, drug resistance and immunotherapy. Cancer Biol Ther 5:741–744
Lyon RC, Cohen JS, Faustino PJ, Megnin F, Myers CE (1988) Glucose metabolism in drug-sensitive and drug-resistant human breast cancer cells monitored by magnetic resonance spectroscopy. Cancer Res 48:870–877
Cao X, Fang L, Gibbs S, Huang Y, Dai Z, Wen P et al (2007) Glucose uptake inhibitor sensitizes cancer cells to daunorubicin and overcomes drug resistance in hypoxia. Cancer Chemother Pharmacol 59:495–505
Lee C, Raffaghello L, Brandhorst S, Safdie FM, Bianchi G, Martin-Montalvo A et al (2012) Fasting cycles retard growth of tumors and sensitize a range of cancer cell types to chemotherapy. Sci Transl Med 4:124ra27
Komurov K, Tseng J-T, Muller M, Seviour EG, Moss TJ, Yang L et al (2012) The glucose-deprivation network counteracts lapatinib-induced toxicity in resistant ErbB2-positive breast cancer cells. Mol Syst Biol 8:596
Sevick EM, Jain RK (1989) Geometric resistance to blood flow in solid tumors perfused ex vivo: effects of tumor size and perfusion pressure. Cancer Res 49:3506–3512
Weiss L, Tveit E, Hultborn R (1985) Vascular resistance characteristics of 7,12-dimethylbenz(a)anthracene-induced rat mammary tumors and normal tissues as studied in vitro. Cancer Res 45:2478–2480
Tveit E, Weiss L, Lundstam S, Hultborn R (1987) Perfusion characteristics and norepinephrine reactivity of human renal carcinoma. Cancer Res 47:4709–4713
Trédan O, Galmarini CM, Patel K, Tannock IF (2007) Drug resistance and the solid tumor microenvironment. J Natl Cancer Inst 99:1441–1454
Salnikov AV, Iversen VV, Koisti M, Sundberg C, Johansson L, Stuhr LB et al (2003) Lowering of tumor interstitial fluid pressure specifically augments efficacy of chemotherapy. FASEB J 17:1756–1758
Késmárky G, Kenyeres P, Rábai M, Tóth K (2008) Plasma viscosity: a forgotten variable. Clin Hemorheol Microcirc 39:243–246
Kwon O, Krishnamoorthy M, Cho YI, Sankovic JM, Banerjee RK (2008) Effect of blood viscosity on oxygen transport in residual stenosed artery following angioplasty. J Biomech Eng 130:011003
Stetler-Stevenson WG, Aznavoorian S, Liotta LA (1993) Tumor cell interactions with the extracellular matrix during invasion and metastasis. Annu Rev Cell Biol 9:541–573
Aoudjit F, Vuori K (2012) Integrin Signaling in Cancer Cell Survival and Chemoresistance. Chemother Res Pract 1–16
Netti PA, Berk DA, Swartz MA, Grodzinsky AJ, Jain RK (2000) Role of extracellular matrix assembly in interstitial transport in solid tumors. Cancer Res 60:2497–2503
Rintoul RC, Sethi T (2001) The role of extracellular matrix in small-cell lung cancer. Lancet Oncol 2:437–442
Rintoul RC, Sethi T (2002) Extracellular matrix regulation of drug resistance in small-cell lung cancer. Clin Sci (Lond) 102:417–424
Naci D, El Azreq M-A, Chetoui N, Lauden L, Sigaux F, Charron D et al (2012) α2β1 integrin promotes chemoresistance against doxorubicin in cancer cells through extracellular signal-regulated kinase (ERK). J Biol Chem 287:17065–17076
Haslam IS, Jones K, Coleman T, Simmons NL (2008) Induction of P-glycoprotein expression and function in human intestinal epithelial cells (T84). Biochem Pharmacol 76:850–861
Schuetz EG, Beck WT, Schuetz JD (1996) Modulators and substrates of P-glycoprotein and cytochrome P4503A coordinately up-regulate these proteins in human colon carcinoma cells. Mol Pharmacol 49:311–318
Yumoto R, Murakami T, Sanemasa M, Nasu R, Nagai J, Takano M (2001) Pharmacokinetic interaction of cytochrome P450 3A-related compounds with rhodamine 123, a P-glycoprotein substrate, in rats pretreated with dexamethasone. Drug Metab Dispos 29:145–151
Aquilante CL, Letrent SP, Pollack GM, Brouwer KL (2000) Increased brain P-glycoprotein in morphine tolerant rats. Life Sci 66:47–51
El Hafny B, Chappey O, Piciotti M, Debray M, Boval B, Roux F (1997) Modulation of P-glycoprotein activity by glial factors and retinoic acid in an immortalized rat brain microvessel endothelial cell line. Neurosci Lett 236:107–111
Dürr D, Stieger B, Kullak-Ublick GA, Rentsch KM, Steinert HC, Meier PJ et al (2000) St John’s Wort induces intestinal P-glycoprotein/MDR1 and intestinal and hepatic CYP3A4. Clin Pharmacol Ther 68:598–604
Jonsson O, Behnam-Motlagh P, Persson M, Henriksson R, Grankvist K (1999) Increase in doxorubicin cytotoxicity by carvedilol inhibition of P-glycoprotein activity. Biochem Pharmacol 58:1801–1806
Anglicheau D, Pallet N, Rabant M, Marquet P, Cassinat B, Méria P et al (2006) Role of P-glycoprotein in cyclosporine cytotoxicity in the cyclosporine-sirolimus interaction. Kidney Int 70:1019–1025
Takara K, Tanigawara Y, Komada F, Nishiguchi K, Sakaeda T, Okumura K (1999) Cellular pharmacokinetic aspects of reversal effect of itraconazole on P-glycoprotein-mediated resistance of anticancer drugs. Biol Pharm Bull 22:1355–1359
Floren LC, Bekersky I, Benet LZ, Mekki Q, Dressler D, Lee JW et al (1997) Tacrolimus oral bioavailability doubles with coadministration of ketoconazole. Clin Pharmacol Ther 62:41–49
Callaghan R, Riordan JR (1993) Synthetic and natural opiates interact with P-glycoprotein in multidrug-resistant cells. J Biol Chem 268:16059–16064
Callaghan R, Higgins CF (1995) Interaction of tamoxifen with the multidrug resistance P-glycoprotein. Br J Cancer 71:294–299
Chen J, Balmaceda C, Bruce JN, Sisti MB, Huang M, Cheung YKK et al (2006) Tamoxifen paradoxically decreases paclitaxel deposition into cerebrospinal fluid of brain tumor patients. J Neurooncol 76:85–92
Jovelet C, Bénard J, Forestier F, Farinotti R, Bidart JM, Gil S (2012) Inhibition of P-glycoprotein functionality by vandetanib may reverse cancer cell resistance to doxorubicin. Eur J Pharm Sci 46:484–491
Xu H-B, Li L, Fu J, Mao X-P, Xu L-Z (2012) Reversion of multidrug resistance in a chemoresistant human breast cancer cell line by β-elemene. Pharmacology 89:303–312
Compliance with ethical guidelines
Competing interests The authors declare that they have no competing interests.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Alfarouk, K.O., Stock, CM., Taylor, S. et al. Resistance to cancer chemotherapy: failure in drug response from ADME to P-gp. Cancer Cell Int 15, 71 (2015). https://doi.org/10.1186/s12935-015-0221-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12935-015-0221-1